
Organometallic chemistry has become a cornerstone of modern organic synthesis. In particular, transition metal-promoted pericyclic reactions figure prominently in many complexity-creating synthetic transformations, their appeal lying in the fact that by using appropriate metal promoters, sluggish or even Woodward-Hoffmann-forbidden processes can be achieved efficiently.
Compared to that of metal-promoted cycloadditions, the synthetic potential of metal-promoted sigmatropic rearrangements has been largely untapped. We are using computational quantum chemistry to predict mechanisms and selectivities for such reactions and our predictions will then be tested experimentally.
Initially, we have focused on the following question: can we find an organometallic group that will promote efficient and stereoselective [1,3]-alkyl shifts? Metal-free [1,3] shifts generally require high temperatures and tend to produce mixtures of products resulting from biradical intermediates. Our goal is to overcome these shortcomings by stabilizing the transition states and/or high energy intermediates involved in thermal [1,3]-alkyl shifts by metal complexation. We are surveying various transition metals and ligands, in pursuit of an organometallic group that will promote this type of reaction in an efficient, general, and stereocontrolled manner.
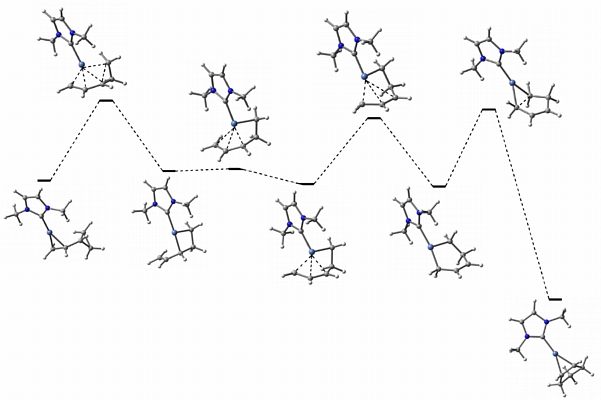
In collaboration with Janis Louie at the University of Utah, we are examining nickel-promoted [1,3]-alkyl shifts of vinylcyclopropanes. The Louie group has shown that nickel/N-heterocyclic carbene complexes can catalyze these reactions at relatively low temperatures. We are examing the mechanisms of these processes computationally (an example of a computed reaction pathway is shown above) with the ultimate goal of designing even more efficient and stereoselective catalyst systems.
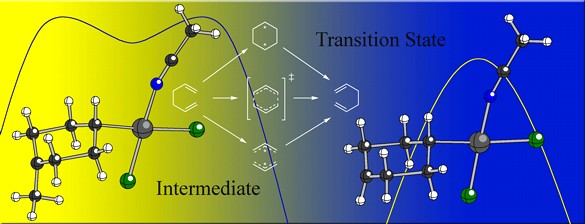
We are also exploring palladium-promoted [3,3]-shifts of 1,5-hexadienes (Cope rearrangements; see below). We've discovered that although the Pd(II) catalyzed [3,3] shift can proceed through a stepwise associative pathway (if appropriately substituted with a cation-stabilizing group), a simple change in substitution position or nature (e.g., an aptly placed electron withdrawing group) makes a concerted pathway energetically favorable. Since the Pd(II) promoted Cope rearrangement, much like its metal-free counterpart, changes mechanism based upon the nature and position of appended substituents, both can be described as "chameleonic".
We continue to explore a variety of pericyclic reactions promoted by transition metals...
Wang, S. C.; Tantillo, D. J. J. Organomet. Chem. 2006, 691, 4386-4392: "Metal Promoted Vinylcyclopropane-Cyclopentene Rearrangements: Reactions Ripe for Mechanism-Based Catalyst Design," invited contribution to special "Theory and Mechanistic Studies" issue.
Siebert, M. R.; Tantillo, D. J. J. Am. Chem. Soc. 2007, 129, 8686-8687: "Transition State Complexation in Palladium-Promoted [3,3] Sigmatropic Shifts"
Wang, S. C.; Troast, D.; Sheridan-Conda, M.; Zuo, G.; LaGarde, D.; Louie, J.; Tantillo, D. J. J. Org. Chem. 2009, 74, 7822-7833: "Mechanism of the Ni(0)-Catalyzed Vinylcyclopropane - Cyclopentene Rearrangement"
Gutierrez, O.; Tantillo, D. J. Organometallics 2010, 29, 3541-3545: "Transition Metal Intervention for a Classic Reaction - Assessing the Feasibility of Ni(0)-promoted [1,3] Sigmatropic Shifts of Bicyclo[3.2.0]hept-2-enes"
Siebert, M. R.; Yudin, A. K.; Tantillo, D. J. Eur. J. Org. Chem. 2011, 553-561: "The Effect of Strain on the Rh(I)-Catalyzed Rearrangement of Allyl Amines"
Hong, Y. J.; Tantillo, D. J. Organometallics 2011, 30, 5825-5831: "Does Gold as a Substituent Accelerate [3,3] Sigmatropic Shifts?"
Gutierrez, O.; Harrison, J. G.; Pemberton, R. P; Tantillo, D. J. Chem. Eur. J. 2012, 18, 11029-11035: "Reexamining the Mechanisms of Competing Pericyclic Reactions of 1,3,7-Octatriene"
Felix, R. J.; Gutierrez, O.; Tantillo, D. J., Gagne, M. R. J. Org. Chem. 2013, 78, 5685-5690: "Gold(I)-Catalyzed Formation of Bicyclo[4.2.0]oct-1-enes"
Gutierrez, O.; Harrison, J. G.; Felix, R. J.; Guzman, F. C.; Gagne, M. R.; Tantillo, D. J. Chem. Sci. 2013, 4, 3894-3898: "Carbonium vs. Carbenium Ion-like Transition State Geometries for Carbocation Cyclization - How Strain Associated with Bridging Affects 5-exo vs. 6-endo Selectivity"
Van Hoveln, R.; Hudson, B. M.; Wedler, H. B.; Bates, D. M.; Le Gros, G.; Tantillo, D. J.; Schomaker, J. M. J. Am. Chem. Soc. 2015, 137, 5346-5354: "Mechanistic Studies of Copper(I)-Catalyzed 1,3-Halogen Migration"
Harrison, J. G.; Gutierrez, O.; Jana, N.; Driver, T. G.; Tantillo, D. J. J. Am. Chem. Soc. 2016, 138, 487-490: "Mechanism of Rh2(II)-Catalyzed Indole Formation: The Catalyst Does Not Control Product Selectivity"
Laconsay, C. J.; Tantillo, D. J. ACS Catal. 2021, 11, 829-839: "Metal-Bound or Free Ylides as Reaction Intermediates in Metal-Catalyzed [2,3]-Sigmatropic Rearrangements? It Depends"
Rayo, D. F. L.; Hong, Y.J.; Campeau, D.; Tantillo, D. J.; Gagosz, F. Chem. Eur. J. 2021, 27, 10637-10648: "On the Mechanism of Au-catalyzed Enynamide-Yne Dehydro-Diels-Alder Reactions: An Experimental and Computational Study"
Ahmed, Y. G.; Tantillo, D. J. Angew. Chem. Int. Ed. 2023, e202300288: "Designing an Apparently Orbital Symmetry Forbidden [3s,5s] Sigmatropic Shift through Transition State Complexation and Stereoelectronic Effects"
Cao, Y.; Kong, W.-Y.; Desnoo, W.; Tantillo, D. J. Eur. J. Org. Chem. 2024, e202301183: "Metallocarbenes as Substituents in 8-Electron Electrocyclizations"
Selina C. Wang and Dean J. Tantillo: "Mechanistic Studies of Nickel Catalyzed Vinylcyclopropane-Cyclopentene Rearrangements." Poster presented by Selina Wang at the Bradford Borge Weekend, University of California, Davis, CA, February 24-25, 2006.
Dean J. Tantillo and Selina C. Wang: "Mechanistic Studies of Nickel Catalyzed Vinylcyclopropane-Cyclopentene Rearrangements." Poster presented by Selina Wang at the 232nd ACS National Meeting, San Francisco, CA, September 10-14, 2006; paper COMP 252.
Matthew R. Siebert and Dean J. Tantillo: "Palladium(II)-Promoted Cope Rearrangement." Poster presented by Matt Siebert at the 232nd ACS National Meeting, San Francisco, CA, September 10-14, 2006; paper ORGN 819.
Selina C. Wang and Dean J. Tantillo: "Mechanistic Studies on Molebdenum-Promoted Sigmatropic Rearrangements of Bicyclo[6.1.0]nona-2,4,6-triene Complexes." Poster presented by Selina Wang at the National Organic Symposium, Durham, NC, June 3-7, 2007; paper D66.
Matthew R. Siebert and Dean J. Tantillo: "Palladium(II)-Promoted Cope Rearrangement." Poster presented by Matt Siebert at the SYLICCO.07 Symposium, Davis, CA, July 26, 2007
Selina C. Wang and Dean J. Tantillo: "Mechanistic Studies on Molebdenum-Promoted Sigmatropic Rearrangements of Bicyclo[6.1.0]nona-2,4,6-triene Complexes." Poster presented by Selina Wang at the SYLICCO.07 Symposium, Davis, CA, July 26, 2007.
Matthew R. Siebert and Dean J. Tantillo: "Quantum Chemical Investigation of Synthetically Useful Rh-Catalyzed Reactions." Poster presented by Matt Siebert at the National Organic Symposium, Boulder, CO, June 7-11, 2009; paper A44
Osvaldo Gutierrez and Dean J. Tantillo: "Engineering an Orbital Forbidden [3s,5s]-Sigmatropic Shift through Palladium(II) Promoted Transition State Complexation." Poster presented by Osvaldo Gutierrez at the 33rd Reaction Mechanisms Conference, University of Massachusetts, Amherst, MA, June 23-26, 2010.